Robust Phylogenetic Inference over Parallel and Distributed Digital Evolution Systems
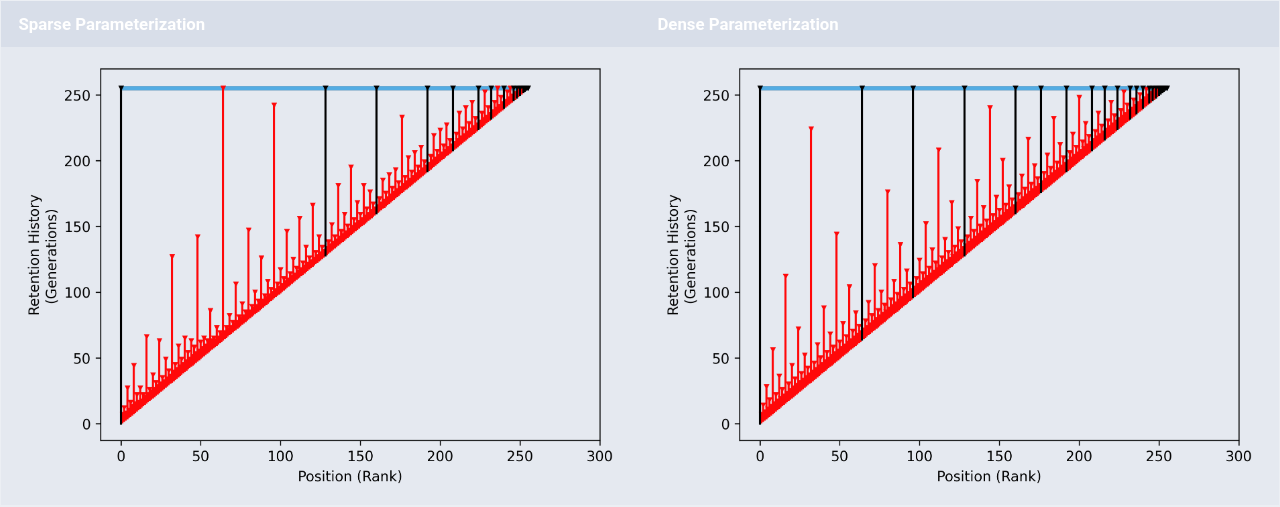 Retention visualization for hereditary stratigraphy policy.
Retention visualization for hereditary stratigraphy policy.
The capability to detect phylogenetic cues within digital evolution has become increasingly necessary in both applied and scientific contexts. These cues unlock post hoc insight into evolutionary history — particularly with respect to ecology and selection pressure — but also can be harnessed to drive digital evolution algorithms as they unfold. However, parallel and distributed evaluation complicates, among other concerns, maintenance of an evolutionary record. Existing phylogenetic record keeping requires inerrant and complete collation of birth and death reports within a centralized data structure. Such perfect tracking approaches are brittle to data loss or corruption and impose communication overhead.
A phylogenetic inference approach, as opposed to phylogenetic tracking, has potential to improve scalability and robustness. Under such a model, history is estimated from comparison of available extant genomes — aligning with the familiar paradigm of phylogenetic work in wet biology. However, this raises the question of how best to design digital genomes to facilitate phylogenetic inference.
This work introduces a new technique, called hereditary stratigraphy, that works by attaching a set of immutable historical “checkpoints” — referred to as strata — as an annotation on evolving genomes.
Checkpoints can be strategically discarded to reduce annotation size at the cost of increasing inference uncertainty.
An accompanying software library, hstrat, provides a plug-and-play implementation of hereditary stratigraphy that can be incorporated into any digital evolution system.
Publications & Software
View at Publisher
| Authors | Connor Yang, Joey Wagner, Emily Dolson, Luis Zaman, Matthew Andres Moreno |
| Date | June 17th, 2025 |
| DOI | 10.48550/arXiv.2506.12975 |
| Venue | arXiv |
Abstract
Due to ongoing accrual over long durations, a defining characteristic of real-world data streams is the requirement for rolling, often real-time, mechanisms to coarsen or summarize stream history. One common data structure for this purpose is the ring buffer, which maintains a running downsample comprising most recent stream data. In some downsampling scenarios, however, it can instead be necessary to maintain data items spanning the entirety of elapsed stream history. Fortunately, approaches generalizing the ring buffer mechanism have been devised to support alternate downsample compositions, while maintaining the ring buffer’s update efficiency and optimal use of memory capacity. The Downstream library implements algorithms supporting three such downsampling generalizations: (1) “steady,” which curates data evenly spaced across the stream history; (2) “stretched,” which prioritizes older data; and (3) “tilted,” which prioritizes recent data. To enable a broad spectrum of applications ranging from embedded devices to high-performance computing nodes and AI/ML hardware accelerators, Downstream supports multiple programming languages, including C++, Rust, Python, Zig, and the Cerebras Software Language. For seamless interoperation, the library incorporates distribution through multiple packaging frameworks, extensive cross-implementation testing, and cross-implementation documentation.
BibTeX
@misc{yang2025downstream,
doi={10.48550/arXiv.2506.12975},
url={https://arxiv.org/abs/2506.12975},
title={Downstream: efficient cross-platform algorithms for fixed-capacity stream downsampling},
author={Connor Yang and Joey Wagner and Emily Dolson and Luis Zaman and Matthew Andres Moreno},
year={2025},
eprint={2506.12975},
archivePrefix={arXiv},
primaryClass={cs.DS},
}
Citation
Yang C., Wagner J., Dolson E., Zaman L., & Moreno M. A. (2025). Downstream: efficient cross-platform algorithms for fixed-capacity stream downsampling. arXiv preprint arXiv:2506.12975. https://doi.org/10.48550/arXiv.2506.12975
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez-Papa, Emily Dolson |
| Date | May 1st, 2025 |
| DOI | 10.1162/artl_a_00470 |
| Venue | Artifical Life |
Abstract
Evolutionary dynamics are shaped by a variety of fundamental, generic drivers, including spatial structure, ecology, and selection pressure. These drivers impact the trajectory of evolution, and have been hypothesized to influence phylogenetic structure. For instance, they can help explain natural history, steer behavior of contemporary evolving populations, and influence efficacy of application-oriented evolutionary optimization. Likewise, in inquiry-oriented artificial life systems, these drivers constitute key building blocks for open-ended evolution. Here, we set out to assess (1) if spatial structure, ecology, and selection pressure leave detectable signatures in phylogenetic structure, (2) the extent, in particular, to which ecology can be detected and discerned in the presence of spatial structure, and (3) the extent to which these phylogenetic signatures generalize across evolutionary systems. To this end, we analyze phylogenies generated by manipulating spatial structure, ecology, and selection pressure within three computational models of varied scope and sophistication. We find that selection pressure, spatial structure, and ecology have characteristic effects on phylogenetic metrics, although these effects are complex and not always intuitive. Signatures have some consistency across systems when using equivalent taxonomic unit definitions (e.g., individual, genotype, species). Further, we find that sufficiently strong ecology can be detected in the presence of spatial structure. We also find that, while low-resolution phylogenetic reconstructions can bias some phylogenetic metrics, high-resolution reconstructions recapitulate them faithfully. Although our results suggest potential for evolutionary inference of spatial structure, ecology, and selection pressure through phylogenetic analysis, further methods development is needed to distinguish these drivers’ phylometric signatures from each other and to appropriately normalize phylogenetic metrics. With such work, phylogenetic analysis could provide a versatile toolkit to study large-scale evolving populations.
BibTeX
@article{moreno2025ecology,
author = {Moreno, Matthew Andres and Rodriguez-Papa, Santiago and Dolson, Emily},
title = {Ecology, Spatial Structure, and Selection Pressure Induce Strong Signatures in Phylogenetic Structure},
journal = {Artificial Life},
volume = {31},
number = {2},
pages = {129-152},
year = {2025},
month = {05},
issn = {1064-5462},
doi = {10.1162/artl_a_00470},
url = {https://doi.org/10.1162/artl\_a\_00470},
eprint = {https://direct.mit.edu/artl/article-pdf/31/2/129/2520922/artl\_a\_00470.pdf},
}
Citation
Matthew Andres Moreno, Santiago Rodriguez-Papa, Emily Dolson; Ecology, Spatial Structure, and Selection Pressure Induce Strong Signatures in Phylogenetic Structure. Artif Life 2025; 31 (2): 129–152. doi: https://doi.org/10.1162/artl_a_00470
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Anika Ranjan, Emily Dolson, Luis Zaman |
| Date | December 5th, 2024 |
| DOI | 10.1109/ALIFE-CIS64968.2025.10979833 |
| Venue | 2025 IEEE Symposium on Computational Intelligence in Artificial Life and Cooperative Intelligent Systems |
Abstract
Computer simulations are an important tool for studying the mechanics of biological evolution. In particular, agent-based approaches provide an opportunity to collect high-quality records of ancestry relationships. Such phylogenies can provide insight into evolutionary dynamics within these simulations. Previous work generally tracks lineages directly, yielding an exact phylogenetic record of evolutionary history. However, challenges exist in scaling direct ancestry-tracking approaches to highly-distributed, many-processor evolution in silico. An alternative approach is to estimate phylogenetic history via non-coding annotations on digital genomes, akin to how bioinformaticians build phylogenies by assessing genetic similarities between organisms. Recent work has extended this “hereditary stratigraphy” approach to support powerful hardware accelerator platforms, such as the Cerebras Wafer-Scale Engine. Although these second-generation “surface”-based hereditary stratigraphy algorithms have demonstrated order-of-magnitude speedups over first-generation “column”-based algorithms, it remains unknown how they impact the accuracy of reconstructed phylogenies. To address this question, we assessed reconstruction accuracy under alternative configurations across a matrix of evolutionary conditions varying in selection pressure, spatial structure, and ecological dynamics. Encouragingly, we find that the second-generation approaches provide higher reconstruction quality across most surveyed conditions.
BibTeX
@inproceedings{moreno2025testing,
author={Moreno, Matthew Andres and Ranjan, Anika and Dolson, Emily and Zaman, Luis},
booktitle={2025 IEEE Symposium on Computational Intelligence in Artificial Life and Cooperative Intelligent Systems (ALIFE-CIS)},
title={Testing the Inference Accuracy of Accelerator-Friendly Approximate Phylogeny Tracking},
year={2025},
pages={1-9},
location = {Trondheim, Norway},
publisher = {IEEE},
address = {Piscataway, NJ, USA},
doi={10.1109/ALIFE-CIS64968.2025.10979833},
url={https://doi.org/10.1109/ALIFE-CIS64968.2025.10979833}
}
Citation
M. A. Moreno, A. Ranjan, E. Dolson and L. Zaman, “Testing the Inference Accuracy of Accelerator-Friendly Approximate Phylogeny Tracking,” 2025 IEEE Symposium on Computational Intelligence in Artificial Life and Cooperative Intelligent Systems (ALIFE-CIS), Trondheim, Norway, 2025, pp. 1-9, doi: 10.1109/ALIFE-CIS64968.2025.10979833.
View at Publisher
| Authors | Matthew Andres Moreno, Connor Yang |
| Date | December 5th, 2024 |
| Venue | Python package published via PyPI |
downstream provides efficient, constant-space implementations of stream curation algorithms for multiple programming languages
View at Publisher
| Authors | Matthew Andres Moreno, Connor Yang, Emily Dolson, Luis Zaman |
| Date | November 16th, 2024 |
| Venue | The International Conference for High Performance Computing, Networking, Storage, and Analysis (SC24) |
Abstract
Emerging ML/AI hardware accelerators, like the 850,000 processor Cerebras Wafer-Scale Engine (WSE), hold great promise to scale up the capabilities of evolutionary computation. However, challenges remain in maintaining visibility into underlying evolutionary processes while efficiently utilizing these platforms’ large processor counts. Here, we focus on the problem of extracting phylogenetic history. We present a tracking-enabled asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations per minute for population sizes reaching 16 million. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction of clear phylometric signals that differentiate adaptive dynamics. Kernel code implementing the island-model GA supports drop-in customization to support any fixed-length genome content and fitness criteria, benefiting further explorations within the evolutionary biology and evolutionary computation communities.
BibTeX
@inproceedings{moreno2024trackable_sc,
author = {Matthew Andres Moreno and Connor Yang and Emily Dolson and Luis Zaman},
title = {Trackable Agent-Based Evolution Models at Wafer Scale},
year = {2024},
url = {https://sc24.supercomputing.org/proceedings/poster/poster_pages/post166.html},
booktitle = {SC24 Research Poster and ACM Student Research Competition Poster Archive},
numpages = {2},
location = {Atlanta, Georgia}
}
Citation
Matthew Andres Moreno, Connor Yang, Emily Dolson, and Luis Zaman. 2024. Trackable Agent-Based Evolution Models at Wafer Scale. In SC24 Research Poster and ACM Student Research Competition Poster Archive. https://sc24.supercomputing.org/proceedings/poster/poster_pages/post166.html
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Mark T. Holder, Jeet Sukumaran |
| Date | September 23rd, 2024 |
| DOI | 10.21105/joss.06943 |
| Venue | Journal of Open Source Software |
Abstract
Contemporary bioinformatics has seen in profound new visibility into the composition, structure, and history of the natural world around us. Arguably, the central pillar of bioinformatics is phylogenetics – the study of hereditary relatedness among organisms. Insight from phylogenetic analysis has touched nearly every corner of biology. Examples range across natural history, population genetics and phylogeography, conservation biology, public health, medicine, in vivo and in silico experimental evolution, application-oriented evolutionary algorithms, and beyond. High-throughput genetic and phenotypic data has realized groundbreaking results, in large part, through conjunction with open-source software used to process and analyze it. Indeed, the preceding decades have ushered in a flourishing ecosystem of bioinformatics software applications and libraries. Over the course of its nearly fifteen-year history, the DendroPy library for phylogenetic computation in Python has established a generalist niche in serving the bioinformatics community. Here, we report on the recent major release of the library, DendroPy version 5. The software release represents a major milestone in transitioning the library to a sustainable long-term development and maintenance trajectory. As such, this work positions DendroPy to continue fulfilling a key supporting role in phyloinformatics infrastructure.
BibTeX
@article{moreno2024dendropy,
doi = {10.21105/joss.06943},
url = {https://doi.org/10.21105/joss.06943},
year = {2024},
publisher = {The Open Journal},
volume = {9},
number = {101},
pages = {6943},
author = {Matthew Andres Moreno and Mark T. Holder and Jeet Sukumaran},
title = {DendroPy 5: a mature Python library for phylogenetic computing},
journal = {Journal of Open Source Software}
}
Citation
Moreno, M. A., Holder, M. T., & Sukumaran, J. (2024). DendroPy 5: a mature Python library for phylogenetic computing. Journal of Open Source Software, 9(101), 6943, https://doi.org/10.21105/joss.06943
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Luis Zaman, Emily Dolson |
| Date | September 10th, 2024 |
| DOI | 10.48550/arXiv.2409.06199 |
| Venue | arXiv |
Abstract
Operations over data streams typically hinge on efficient mechanisms to aggregate or summarize history on a rolling basis. For high-volume data steams, it is critical to manage state in a manner that is fast and memory efficient — particularly in resource-constrained or real-time contexts. Here, we address the problem of extracting a fixed-capacity, rolling subsample from a data stream. Specifically, we explore “data stream curation” strategies to fulfill requirements on the composition of sample time points retained. Our “DStream” suite of algorithms targets three temporal coverage criteria: (1) steady coverage, where retained samples should spread evenly across elapsed data stream history; (2) stretched coverage, where early data items should be proportionally favored; and (3) tilted coverage, where recent data items should be proportionally favored. For each algorithm, we prove worst-case bounds on rolling coverage quality. We focus on the more practical, application-driven case of maximizing coverage quality given a fixed memory capacity. As a core simplifying assumption, we restrict algorithm design to a single update operation: writing from the data stream to a calculated buffer site — with data never being read back, no metadata stored (e.g., sample timestamps), and data eviction occurring only implicitly via overwrite. Drawing only on primitive, low-level operations and ensuring full, overhead-free use of available memory, this “DStream” framework ideally suits domains that are resource-constrained, performance-critical, and fine-grained (e.g., individual data items as small as single bits or bytes). The proposed approach supports O(1) data ingestion via concise bit-level operations. To further practical applications, we provide plug-and-play open-source implementations targeting both scripted and compiled application domains.
BibTeX
@misc{moreno2024structured,
doi={10.48550/arXiv.2409.06199},
url={https://arxiv.org/abs/2409.06199},
title={Structured Downsampling for Fast, Memory-efficient Curation of Online Data Streams},
author={Matthew Andres Moreno and Luis Zaman and Emily Dolson},
year={2024},
eprint={2409.06199},
archivePrefix={arXiv},
primaryClass={cs.DS}
}
Citation
Moreno M. A., Zaman L., & Dolson E. (2024). Structured Downsampling for Fast, Memory-efficient Curation of Online Data Streams. arXiv preprint arXiv:2409.06199. https://doi.org/10.48550/arXiv.2409.06199
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Anika Ranjan, Emily Dolson, Luis Zaman |
| Date | May 16th, 2024 |
| DOI | 10.48550/arXiv.2405.10183 |
| Venue | arXiv |
Abstract
Computer simulations are an important tool for studying the mechanics of biological evolution. In particular, in silico work with agent-based models provides an opportunity to collect high-quality records of ancestry relationships among simulated agents. Such phylogenies can provide insight into evolutionary dynamics within these simulations. Existing work generally tracks lineages directly, yielding an exact phylogenetic record of evolutionary history. However, direct tracking can be inefficient for large-scale, many-processor evolutionary simulations. An alternate approach to extracting phylogenetic information from simulation that scales more favorably is post hoc estimation, akin to how bioinformaticians build phylogenies by assessing genetic similarities between organisms. Recently introduced “hereditary stratigraphy” algorithms provide means for efficient inference of phylogenetic history from non-coding annotations on simulated organisms’ genomes. A number of options exist in configuring hereditary stratigraphy methodology, but no work has yet tested how they impact reconstruction quality. To address this question, we surveyed reconstruction accuracy under alternate configurations across a matrix of evolutionary conditions varying in selection pressure, spatial structure, and ecological dynamics. We synthesize results from these experiments to suggest a prescriptive system of best practices for work with hereditary stratigraphy, ultimately guiding researchers in choosing appropriate instrumentation for large-scale simulation studies.
BibTeX
@misc{moreno2024guide,
doi={10.48550/arXiv.2405.10183},
url={https://arxiv.org/abs/2405.10183},
title={A Guide to Tracking Phylogenies in Parallel and Distributed Agent-based Evolution Models},
author={Matthew Andres Moreno and Anika Ranjan and Emily Dolson and Luis Zaman},
year={2024},
eprint={2405.10183},
archivePrefix={arXiv},
primaryClass={cs.NE}
}
Citation
Moreno, M. A., Ranjan, A., Dolson, E., & Zaman, L. (2024). A Guide to Tracking Phylogenies in Parallel and Distributed Agent-based Evolution Models. arXiv preprint arXiv:2405.10183. https://doi.org/10.48550/arXiv.2405.10183
Supporting Materials
View at Publisher
| Authors | Emily Dolson, Santiago Rodriguez-Papa, Matthew Andres Moreno |
| Date | May 15th, 2024 |
| DOI | 10.48550/arXiv.2405.09389 |
| Venue | arXiv |
Abstract
In silico evolution instantiates the processes of heredity, variation, and differential reproductive success (the three “ingredients” for evolution by natural selection) within digital populations of computational agents. Consequently, these populations undergo evolution, and can be used as virtual model systems for studying evolutionary dynamics. This experimental paradigm — used across biological modeling, artificial life, and evolutionary computation — complements research done using in vitro and in vivo systems by enabling experiments that would be impossible in the lab or field. One key benefit is complete, exact observability. For example, it is possible to perfectly record all parent-child relationships across simulation history, yielding complete phylogenies (ancestry trees). This information reveals when traits were gained or lost, and also facilitates inference of underlying evolutionary dynamics.
The Phylotrack project provides libraries for tracking and analyzing phylogenies in in silico evolution. The project is composed of 1) Phylotracklib: a header-only C++ library, developed under the umbrella of the Empirical project, and 2) Phylotrackpy: a Python wrapper around Phylotracklib, created with Pybind11. Both components supply a public-facing API to attach phylogenetic tracking to digital evolution systems, as well as a stand-alone interface for measuring a variety of popular phylogenetic topology metrics. Underlying design and C++ implementation prioritizes efficiency, allowing for fast generational turnover for agent populations numbering in the tens of thousands. Several explicit features (e.g., phylogeny pruning and abstraction, etc.) are provided for reducing the memory footprint of phylogenetic information.
BibTeX
@misc{dolson2024phylotrack,
doi={10.48550/arXiv.2405.09389},
url={https://arxiv.org/abs/2405.09389},
title={Phylotrack: C++ and Python libraries for in silico phylogenetic tracking},
author={Emily Dolson and Santiago Rodriguez-Papa and Matthew Andres Moreno},
year={2024},
eprint={2405.09389},
archivePrefix={arXiv},
primaryClass={q-bio.PE}
}
Citation
Dolson, E., Rodriguez-Papa, S., & Moreno, M. A. (2024). Phylotrack: C++ and Python libraries for in silico phylogenetic tracking. arXiv preprint arXiv:2405.09389. https://doi.org/10.48550/arXiv.2405.09389
View at Publisher
| Authors | Matthew Andres Moreno, Connor Yang, Emily Dolson, Luis Zaman |
| Date | May 6th, 2024 |
| DOI | 10.1145/3638530.3664090 |
| Venue | The Genetic and Evolutionary Computation Conference |
Abstract
Emerging ML/AI hardware accelerators, like the 850,000 processor Cerebras Wafer-Scale Engine (WSE), hold great promise to scale up the capabilities of evolutionary computation. However, challenges remain in maintaining visibility into underlying evolutionary processes while efficiently utilizing these platforms’ large processor counts. Here, we focus on the problem of extracting phylogenetic information from digital evolution on the WSE platform. We present a tracking-enabled asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations a minute for population sizes reaching 16 million. This pace enables quadrillions of evaluations a day. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction of clear phylometric signals that differentiate wafer-scale runs with adaptive dynamics enabled versus disabled. Together, these benchmark and validation trials reflect strong potential for highly scalable evolutionary computation that is both efficient and observable. Kernel code implementing the island-model GA supports drop-in customization to support any fixed-length genome content and fitness criteria, allowing it to be leveraged to advance research interests across the community.
BibTeX
@inproceedings{moreno2024trackable_gecco,
author = {Matthew Andres Moreno and Connor Yang and Emily Dolson and Luis Zaman},
title = {Trackable Island-model Genetic Algorithms at Wafer Scale},
pages = {101-102},
isbn = {9798400704956},
year = {2024},
publisher= {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3638530.3664090},
doi = {10.1145/3638530.3664090},
booktitle= {Proceedings of the Genetic and Evolutionary Computation Conference Companion},
numpages = {2},
location = {Melbourne, VIC, Australia},
series = {GECCO '24}
}
Citation
Matthew Andres Moreno, Connor Yang, Emily Dolson, and Luis Zaman. 2024. Trackable Island-model Genetic Algorithms at Wafer Scale. In Proceedings of the Companion Conference on Genetic and Evolutionary Computation (GECCO ‘24 Companion). Association for Computing Machinery, New York, NY, USA. https://doi.org/10.1145/3638530.3664090
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Connor Yang, Emily Dolson, Luis Zaman |
| Date | April 16th, 2024 |
| DOI | 10.1162/isal_a_00830 |
| Venue | The 2024 Conference on Artificial Life |
Abstract
Continuing improvements in computing hardware are poised to transform capabilities for in silico modeling of cross-scale phenomena underlying major open questions in evolutionary biology and artificial life, such as transitions in individuality, eco-evolutionary dynamics, and rare evolutionary events. Emerging ML/AI-oriented hardware accelerators, like the 850,000 processor Cerebras Wafer Scale Engine (WSE), hold particular promise. However, practical challenges remain in conducting informative evolution experiments that efficiently utilize these platforms’ large processor counts. Here, we focus on the problem of extracting phylogenetic information from agent-based evolution on the WSE platform. This goal drove significant refinements to decentralized in silico phylogenetic tracking, reported here. These improvements yield order-of-magnitude performance improvements. We also present an asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations a minute for population sizes reaching 16 million agents. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction, from wafer-scale simulation, of clear phylometric signals that differentiate runs with adaptive dynamics enabled versus disabled. Together, these benchmark and validation trials reflect strong potential for highly scalable agent-based evolution simulation that is both efficient and observable. Developed capabilities will bring entirely new classes of previously intractable research questions within reach, benefiting further explorations within the evolutionary biology and artificial life communities across a variety of emerging high-performance computing platforms.
BibTeX
@inproceedings{moreno2024trackable,
author = {Matthew Andres Moreno and Connor Yang and Emily Dolson and Luis Zaman},
title = {Trackable Agent-based Evolution Models at Wafer Scale},
booktitle = {The 2024 Conference on Artificial Life},
collection = {ALIFE 2024},
publisher = {MIT Press},
year = {2024},
month = {07},
doi={10.1162/isal_a_00830},
url={https://doi.org/10.1162/isal_a_00830},
numpages={12},
pages={87-98},
}
Citation
Moreno, M. A., Yang, C., Dolson, E., & Zaman, L. (2024). Trackable Agent-based Evolution Models at Wafer Scale. In The 2024 Conference on Artificial Life. MIT Press. https://doi.org/10.1162/isal_a_00830
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Emily Dolson |
| Date | March 3rd, 2024 |
| DOI | 10.48550/arXiv.2403.00246 |
| Venue | arXiv |
Abstract
Since the advent of modern bioinformatics, the challenging, multifaceted problem of reconstructing phylogenetic history from biological sequences has hatched perennial statistical and algorithmic innovation. Studies of the phylogenetic dynamics of digital, agent-based evolutionary models motivate a peculiar converse question: how to best engineer tracking to facilitate fast, accurate, and memory-efficient lineage reconstructions? Here, we formally describe procedures for phylogenetic analysis in both serial and distributed computing scenarios. With respect to the former, we demonstrate reference-counting-based pruning of extinct lineages. For the latter, we introduce a trie-based phylogenetic reconstruction approach for “hereditary stratigraphy” genome annotations. This process allows phylogenetic relationships between genomes to be inferred by comparing their similarities, akin to reconstruction of natural history from biological DNA sequences. Phylogenetic analysis capabilities significantly advance distributed agent-based simulations as a tool for evolutionary research, and also benefit application-oriented evolutionary computing. Such tracing could extend also to other digital artifacts that proliferate through replication, like digital media and computer viruses.
BibTeX
@misc{moreno2024analysis,
doi={10.48550/arXiv.2403.00246},
url={https://arxiv.org/abs/2403.00246},
title={Analysis of Phylogeny Tracking Algorithms for Serial and Multiprocess Applications},
author={Matthew Andres Moreno and Santiago {Rodriguez Papa} and Emily Dolson},
year={2024},
eprint={2403.00246},
archivePrefix={arXiv},
primaryClass={cs.DS}
}
Citation
Moreno, M. A., Rodriguez Papa, S., & Dolson, E. (2024). Analysis of Phylogeny Tracking Algorithms for Serial and Multiprocess Applications. arXiv preprint arXiv:2403.00246 https://doi.org/10.48550/arXiv.2403.00246
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Emily Dolson |
| Date | March 3rd, 2024 |
| DOI | 10.48550/arXiv.2403.00266 |
| Venue | arXiv |
Abstract
Data stream algorithms tackle operations on high-volume sequences of read-once data items. Data stream scenarios include inherently real-time systems like sensor networks and financial markets. They also arise in purely-computational scenarios like ordered traversal of big data or long-running iterative simulations. In this work, we develop methods to maintain running archives of stream data that are temporally representative, a task we call “stream curation.” Our approach contributes to rich existing literature on data stream binning, which we extend by providing stateless (i.e., non-iterative) curation schemes that enable key optimizations to trim archive storage overhead and streamline processing of incoming observations. We also broaden support to cover new trade-offs between curated archive size and temporal coverage. We present a suite of five stream curation algorithms that span O(n), O(logn), and O(1) orders of growth for retained data items. Within each order of growth, algorithms are provided to maintain even coverage across history or bias coverage toward more recent time points. More broadly, memory-efficient stream curation can boost the data stream mining capabilities of low-grade hardware in roles such as sensor nodes and data logging devices.
BibTeX
@misc{moreno2024algorithms,
doi={10.48550/arXiv.2403.00266},
url={https://arxiv.org/abs/2403.00246},
title={Algorithms for Efficient, Compact Online Data Stream Curation},
author={Matthew Andres Moreno and Santiago {Rodriguez Papa} and Emily Dolson},
year={2024},
eprint={2403.00266},
archivePrefix={arXiv},
primaryClass={cs.DS}
}
Citation
Moreno, M. A., Rodriguez Papa, S., & Dolson, E. (2024). Algorithms for Efficient, Compact Online Data Stream Curation. arXiv preprint arXiv:2403.00266. https://doi.org/10.48550/arXiv.2403.00266
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | February 18th, 2024 |
| Venue | Genetic Programming Theory and Practice XX |
Abstract
The structure of relatedness among members of an evolved population tells much of its evolutionary history. In application-oriented evolutionary computation (EC), such phylogenetic information can guide algorithm selection and tuning. Although traditional direct tracking approaches provide the perfect phylogenetic record, sexual recombination complicates management and analysis of this data. Taking inspiration from biological science, this work explores a reconstruction-based approach that uses end-state genetic information to estimate phylogenetic history after the fact. We apply recently-developed “hereditary stratigraphy” genome annotations to lineages with sexual recombination to design devices germane to species phylogenies and gene trees. As shown through a series of validation experiments, proposed instrumentation can discern genealogical history, population size changes, and selective sweeps. Fully decentralized by nature, these methods afford new observability at scale, in particular, for distributed EC systems. Such capabilities anticipate continued growth of computational resources available to EC. Accompanying open source software aims to expedite application of reconstruction-based phylogenetic analysis where pertinent.
BibTeX
@incollection{moreno2024methods,
author = {Moreno, Matthew Andres},
editor = {Winkler, Stephan
and Trujillo, Leonardo
and Ofria, Charles
and Hu, Ting},
title = {Methods for Rich Phylogenetic Inference Over Distributed Sexual Populations},
booktitle = {Genetic Programming Theory and Practice XX},
year = 2024,
pages = {125--141},
publisher = {Springer International Publishing},
isbn = {978-981-99-8413-8},
doi = {10.1007/978-981-99-8413-8_7},
url = {https://doi.org/10.1007/978-981-99-8413-8_7},
}
Citation
Moreno, M.A. (2024). Methods for Rich Phylogenetic Inference Over Distributed Sexual Populations. In: Winkler, S., Trujillo, L., Ofria, C., Hu, T. (eds) Genetic Programming Theory and Practice XX. Genetic and Evolutionary Computation. Springer, Singapore. https://doi.org/10.1007/978-981-99-8413-8_7
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Santiago Rodriguez-Papa |
| Date | July 24th, 2023 |
| DOI | 10.1162/isal_a_00694 |
| Venue | The 2023 Conference on Artificial Life |
Abstract
As digital evolution systems grow in scale and complexity, observing and interpreting their evolutionary dynamics will become increasingly challenging. Distributed and parallel computing, in particular, introduce obstacles to maintaining the high level of observability that makes digital evolution a powerful experimental tool. Phylogenetic analyses represent a promising tool for drawing inferences from digital evolution experiments at scale. Recent work has introduced promising techniques for decentralized phylogenetic inference in parallel and distributed digital evolution systems. However, foundational phylogenetic theory necessary to apply these techniques to characterize evolutionary dynamics is lacking. Here, we lay the groundwork for practical applications of distributed phylogenetic tracking in three ways: 1) we present an improved technique for reconstructing phylogenies from tunably-precise genome annotations, 2) we begin the process of identifying how the signatures of various evolutionary dynamics manifest in phylogenetic metrics, and 3) we quantify the impact of reconstruction-induced imprecision on phylogenetic metrics. We find that selection pressure, spatial structure, and ecology have distinct effects on phylogenetic metrics, although these effects are complex and not always intuitive. We also find that, while low-resolution phylogenetic reconstructions can bias some phylogenetic metrics, high-resolution reconstructions recapitulate them faithfully.
BibTeX
@inproceedings{moreno2023toward,
author = {Moreno, Matthew Andres and Dolson, Emily and Rodriguez-Papa, Santiago},
title = {Toward Phylogenetic Inference of Evolutionary Dynamics at Scale},
booktitle = {The 2023 Conference on Artificial Life},
collection = {ALIFE 2023},
publisher = {MIT Press},
pages = {568-668},
year = {2023},
month = {07},
doi = {10.1162/isal_a_00694},
url = {https://doi.org/10.1162/isal\_a\_00694},
eprint = {https://direct.mit.edu/isal/proceedings-pdf/isal/35/79/2149068/isal\_a\_00694.pdf},
}
Citation
Moreno, M. A., Dolson, E., & Rodriguez-Papa, S. (2023). Toward Phylogenetic Inference of Evolutionary Dynamics at Scale. In The 2023 Conference on Artificial Life. MIT Press. https://doi.org/10.1162/isal_a_00694
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | November 7th, 2022 |
| DOI | 10.21105/joss.04866 |
| Venue | Journal of Open Source Software |
Abstract
Digital evolution systems instantiate evolutionary processes over populations of virtual agents in silico. These programs can serve as rich experimental model systems. Insights from digital evolution experiments expand evolutionary theory, and can often directly improve heuristic optimization techniques . Perfect observability, in particular, enables in silico experiments that would be otherwise impossible in vitro or in vivo. Notably, availability of the full evolutionary history (phylogeny) of a given population enables very powerful analyses.
As a slow but highly parallelizable process, digital evolution will benefit greatly by continuing to capitalize on profound advances in parallel and distributed computing, particularly emerging unconventional computing architectures. However, scaling up digital evolution presents many challenges. Among these is the existing centralized perfect-tracking phylogenetic data collection model, which is inefficient and difficult to realize in parallel and distributed contexts. Here, we implement an alternative approach to tracking phylogenies across vast and potentially unreliable hardware networks.
BibTeX
@article{moreno2022hstrat,
doi = {10.21105/joss.04866},
url = {https://doi.org/10.21105/joss.04866},
year = {2022},
publisher = {The Open Journal},
volume = {7},
number = {80},
pages = {4866},
author = {Matthew Andres Moreno and Emily Dolson and Charles Ofria},
title = {hstrat: a Python Package for phylogenetic inference on distributed digital evolution populations},
journal = {Journal of Open Source Software}
}
Citation
Moreno M.A., Dolson, E., & Ofria, C. (2022). hstrat: a Python Package for phylogenetic inference on distributed digital evolution populations. Journal of Open Source Software, 7(80), 4866, https://doi.org/10.21105/joss.04866
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | May 13th, 2022 |
| DOI | 10.1145/3520304.3533937 |
| Venue | The Genetic and Evolutionary Computation Conference |
Abstract
Phylogenetic analyses can also enable insight into evolutionary and ecological dynamics such as selection pressure and frequency dependent selection in digital evolution systems. Traditionally digital evolution systems have recorded data for phylogenetic analyses through perfect tracking where each birth event is recorded in a centralized data structures. This approach, however, does not easily scale to distributed computing environments where evolutionary individuals may migrate between a large number of disjoint processing elements. To provide for phylogenetic analyses in these environments, we propose an approach to infer phylogenies via heritable genetic annotations rather than directly track them. We introduce a “hereditary stratigraphy” algorithm that enables efficient, accurate phylogenetic reconstruction with tunable, explicit trade-offs between annotation memory footprint and reconstruction accuracy. This approach can estimate, for example, MRCA generation of two genomes within 10% relative error with 95% confidence up to a depth of a trillion generations with genome annotations smaller than a kilobyte. We also simulate inference over known lineages, recovering up to 85.70% of the information contained in the original tree using a 64-bit annotation.
BibTeX
@inproceedings{moreno2022hereditary_gecco,
author = {Moreno, Matthew Andres and Dolson, Emily and Ofria, Charles},
title = {Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations},
year = {2022},
isbn = {9781450392686},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3520304.3533937},
doi = {10.1145/3520304.3533937},
booktitle = {Proceedings of the Genetic and Evolutionary Computation Conference Companion},
pages = {65–66},
numpages = {2},
keywords = {phylogenetics, decentralized algorithms, genetic algorithms, digital evolution, genetic programming},
location = {Boston, Massachusetts},
series = {GECCO '22}
}
Citation
Matthew Andres Moreno, Emily Dolson, and Charles Ofria. 2022. Hereditary stratigraphy: genome annotations to enable phylogenetic inference over distributed populations. In Proceedings of the Genetic and Evolutionary Computation Conference Companion (GECCO ‘22). Association for Computing Machinery, New York, NY, USA, 65–66. https://doi.org/10.1145/3520304.3533937
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | May 13th, 2022 |
| DOI | 10.1162/isal_a_00550 |
| Venue | The 2022 Conference on Artificial Life |
Abstract
Phylogenies provide direct accounts of the evolutionary trajectories behind evolved artifacts in genetic algorithm and artificial life systems. Phylogenetic analyses can also enable insight into evolutionary and ecological dynamics such as selection pressure and frequency-dependent selection. Traditionally, digital evolution systems have recorded data for phylogenetic analyses through perfect tracking where each birth event is recorded in a centralized data structure. This approach, however, does not easily scale to distributed computing environments where evolutionary individuals may migrate between a large number of disjoint processing elements. To provide for phylogenetic analyses in these environments, we propose an approach to enable phylogenies to be inferred via heritable genetic annotations rather than directly tracked. We introduce a “hereditary stratigraphy” algorithm that enables efficient, accurate phylogenetic reconstruction with tunable, explicit trade-offs between annotation memory footprint and reconstruction accuracy. In particular, we demonstrate an approach that enables estimation of the most recent common ancestor (MRCA) between two individuals with fixed relative accuracy irrespective of lineage depth while only requiring logarithmic annotation space complexity with respect to lineage depth This approach can estimate, for example, MRCA generation of two genomes within 10% relative error with 95% confidence up to a depth of a trillion generations with genome annotations smaller than a kilobyte. We also simulate inference over known lineages, recovering up to 85.70% of the information contained in the original tree using 64-bit annotations.
BibTeX
@inproceedings{moreno2022hereditary,
author = {Moreno, Matthew Andres and Dolson, Emily and Ofria, Charles},
title = {Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations},
booktitle = {The 2022 Conference on Artificial Life},
collection = {ALIFE 2022},
year = {2022},
month = {07},
doi = {10.1162/isal_a_00550},
url = {https://doi.org/10.1162/isal\_a\_00550},
pages = {418-428},
eprint = {https://direct.mit.edu/isal/proceedings-pdf/isal/34/64/2035363/isal\_a\_00550.pdf},
}
Citation
Moreno, M. A., Dolson, E., & Ofria, C. (2022). Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations. In The 2022 Conference on Artificial Life. MIT Press. https://doi.org/10.1162/isal_a_00550
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
hstrat enables phylogenetic inference on distributed digital evolution populations.
BibTeX
@article{moreno2022hstrat,
doi = {10.21105/joss.04866},
url = {https://doi.org/10.21105/joss.04866},
year = {2022},
publisher = {The Open Journal},
volume = {7},
number = {80},
pages = {4866},
author = {Matthew Andres Moreno and Emily Dolson and Charles Ofria},
title = {hstrat: a Python Package for phylogenetic inference on distributed digital evolution populations},
journal = {Journal of Open Source Software}
}
Citation
Moreno M.A., Dolson, E., & Ofria, C. (2022). hstrat: a Python Package for phylogenetic inference on distributed digital evolution populations. Journal of Open Source Software, 7(80), 4866, https://doi.org/10.21105/joss.04866