
Performance of Genome Class when Using Change Log
🔗 Introduction
For a short introduction about MABE (Modular Agent-Based Evolution platform) as well as the description and performance analysis of other approaches see this post.
🔗 Genome Class
Genome is a sequence of sites that contain heritable and mutable data. Genome is used by other MABE modules, for example as a source of data necessary to construct a brain in MABE.
In biology, a genome is a sequence of four types of nucleotides (A, C, G, T). In MABE, the genome data could be of any type, and for this project the data in our genome will be of std::byte type.
The genome class interface provides several mutation methods, which are used to create an offspring genome from a parent genome, specifically:
- Overwrite mutation - the value at one or more sites is overwritten by a different value
- Insert mutation - one or more sites are inserted into the genome
- Remove mutation - one or more sites are removed from the genome
🔗 Naive Implementation
Genome is a sequence of sites with specific values, e.g.:

It can be naively implemented as a std::vector data structure from the standard library.
In this naive design, all mutations can be implemented using the standard library algorithms on std::vector, specifically:
// A genome that consists of three sites with values 1, 2 and 3
std::vector<std::byte> sites{std::byte(1), std::byte(2), std::byte(3)};
// Overwrite mutation: overwrite using values from segment starting at index
void overwrite(size_t index, const std::vector<std::byte>& segment) {
for (size_t i(0); i < segment.size(); i++) {
sites[index + i] = segment[i];
}
}
// Insert mutation: a segment is inserted at index
void insert(size_t index, const std::vector<std::byte>& segment) {
sites.insert(sites.begin() + index, segment.begin(), segment.end());
}
// Remove mutation: segmentSize sites are removed starting at index
void remove(size_t index, size_t segmentSize) {
sites.erase(sites.begin() + index, sites.begin() + index + segmentSize);
}
The advantages of this approach include:
- All the sites are in contiguous memory -> fast operations due to cache-friendliness (e.g. random access or iterations)
- Use of C++ standard library data structures -> code is simple, readable, expressive and optimized for performance
However, there are some disadvantages:
- Every generation, the whole genome (the whole
sitesvector) is copied and then the mutations are applied to it. In a common situation of a large genome and low mutation rates, it means copying a lot of values that do not change between the parent and the offspring genomes - The
insert()anderase()algorithms have linear time complexity -> inefficient time
The goal of this project was to investigate if storing only the mutations (as opposed to storing the whole genome) for each offspring would provide a better time and memory performance.
🔗 Optimized Implementation Using Change Log
One of the ways to improve the time complexity as well as optimize for memory use is to have a change log. The change log will keep track of the mutations that occurred between the parent and the offsprings over generations. This means only storing the differences between parent genome and it’s offsprings as opposed to storing the whole genome for every offspring.
As we’ve seen above, the genome class has to support the following mutations:
- Overwrite
- Insert
- Remove
My implementation consists of two maps, which, for each offspring genome, store all the necessary information on how this genome is different from the parent:
-
change_log is implemented as a
std::mapand contains the information about the number of inserted and removed sites (a shift in sites compared to the parent genome). It is used to calculate the relationship between a particular site in the offspring genome and the parent genome -
segments_log is implemented as a
std::unordered_mapand stores the segments that were inserted into the genome during mutations

Each genome will have it’s own change_log and segments_log, which in combination with the parent genome will allow the random access to any value in the offspring genome as well as the reconstruction of complete offspring genome (or a part of it) as a contiguous memory block of the necessary sites.
One important detail about the change_log is that it doesn’t store every removed or inserted index, instead each key in the change_log represents all the keys in the range from the current key until the next key (see example below).
For example, a change_log with entries {{0 : 0}, {3 : -2}, {5 : 3}} corresponds to the following mapping:

To access any index, the following code can be used:
--change_log.upper_bound(index); // "upper_bound" returns the key, which is higher than the index, "--" moves to the previous key
In the case of the change_log above, --change_log.upper_bound(7); will return the iterator to the key = 5.
🔗 Remove mutation 
When one or more sites are removed from the genome, a new element {key : value} is added in the change_log map: the key corresponds to the index, at which the remove mutation starts and the value corresponds to the number of sites that were removed.
The following animation shows how the remove mutation is stored in the change_log and how the change_log is then used to reconstruct the offspring genome:

In the animation above, the remove(3, 2) method is called, which corresponds to removing two sites at index 3. The index and the number of removed sites are stored in the change_log as a {key : value} pair.
The change_log can then be used to reconstruct the offspring genome, specifically, the values at indices < 3 in the offspring genome will be the same as in the parent genome. And the sites at the indices >= 3 will be shifted to the left by two sites.
Therefore, in order to access the corresponding values, we need to shift two sites to the right in the parent genome:
offspring[index] = parent[index + 2]
In the change_log map, each value is the the accumulation of all the changes up to the corresponding key, for example, if two elements were removed at index 3 and then three elements were removed at index 5, the accumulated shift at index >= 5 will be -5:

Using this change_log and the parent genome, it is possible to reconstruct the offspring genome by calculating a specific index in the offspring genome in relationship to the parent genome, i.e. for indices:
- < 3: same value as in the parent genome
- >= 3 && <5:
offspring[index] = parent[index + 2] - >= 5:
offspring[index] = parent[index + 5]
🔗 Insert mutation 
Up to now each value in the change_log corresponded to the shift of genome sites relative to its parent genome. The values of the newly inserted sites do not have any relation to the parent genome, therefore, the values for such keys are set to zero. In order to not confuse it with zero sites shift, an additional variable is added to the map: a boolean, which specifies whether sites were inserted at this key:
{key : {val : insert}} // insert == true if sites were inserted at this key
The animation shows an example, where our previous change_log is updated with an insertion of three elements {20, 21, 22} at index 6:

In addition to change_log, we now also use segments_log to store the inserted segment. The std::unordered_map allows constant time access by key.
After the insert mutation is added to the change_log: {6, {0, true}} and segments_log: {6, {20, 21, 22}}, we also add an additional element to know where the inserted segment ends.
Specifically, in this case the element is {9, {-2, false}}, where the key corresponds to the (insert index + segment size): 9 = 6 + 3 and value corresponds to the sites shift up to now (remove 2 sites && remove 3 sites && insert 3 sites): -2 = -2 + (-3) + 3.
Using this change_log and the parent genome, it is possible to reconstruct the offspring genome by calculating a specific index in the offspring genome in relationship to the parent genome, i.e. for indices:
- < 3: same value as in the parent genome
- >= 3 && ind < 5:
offspring[index] = parent[index + 2] - >= 5 && ind < 6:
offspring[index] = parent[index + 5] - >= 6 && ind < 9: segment from
segments_log.at(6) - > 9:
offspring[index] = parent[index + 2]
🔗 All mutations combined
The algorithms for overwrite(), insert() and remove(), are designed in a way to keep the change_log and insertions_log correct after every mutation, so that we can have random access to a genome site and can reconstruct a whole offspring genome or its part.
To sum up, the change log consists of two data structures:
- change_log: keeps track of shifts in sites when remove or insert mutation happens
- Implemented as
std::map: keeping the keys in sorted order allows updating only the keys larger than current key; accessing the key takes logarithmic time - segments_log: stores inserted segments
-
std::unordered_map: accessing a segment by key takes constant time
🔗 Algorithm performance
The plots below show the performance of my approach compared to the naive one for the overwrite(), remove() and insert() mutations methods as well as multi-mutation, when all three methods are applied together.
The performance is shown for a number of genome sizes, specifically (each site corresponds to one byte) 5kB, 20kB, 50kB, 75kB, 100kB, 250kB and 500kB. The mutation rate is 0.1%, which corresponds to the 5, 20, 50, 75, 100, 250 and 500 mutations respectively for each genome size.
The performance of the overwrite() mutation of my approach is very similar to the naive approach:
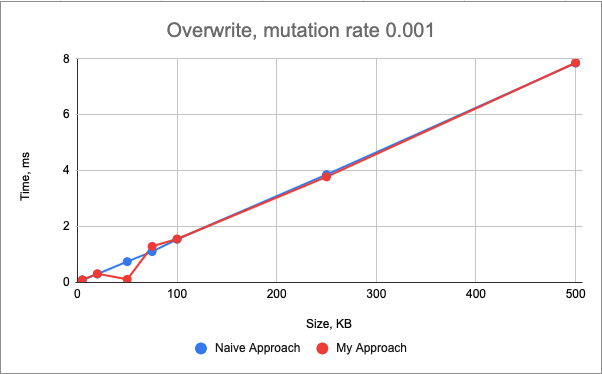
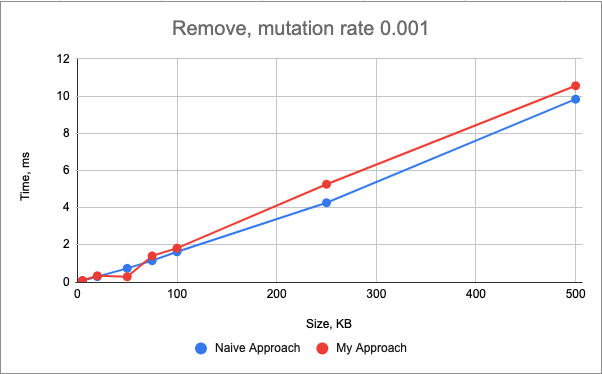
The performance of the remove() mutation is very similar between two approaches too.
In the naive approach, this mutation has linear time complexity relative to the genome size. In my approach, the time complexity is linear with the change_log size (as I need to update all the keys, which follow the current key).
Normally, the genome would be much larger than the change_log, therefore, I would expect my approach to be faster.
However, the std::vector data structure stores the values in the contiguous memory (as opposed to std::map data structure), which results in faster iteration due to the utilization of cache-friendliness.
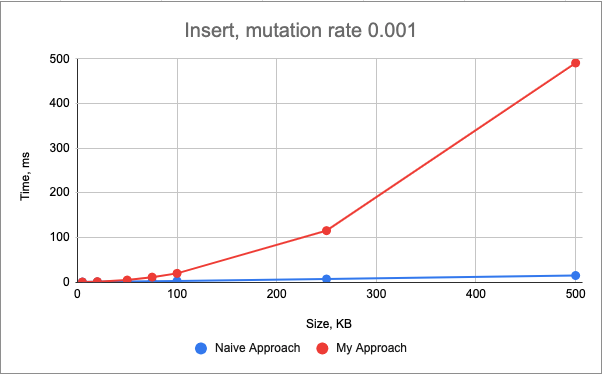
The performance of the insert() mutation is very similar between two approaches for the smaller genomes (< 100kB), however, as the size of the genome increases, the plots start to diverge, showing the strengths of the standard library.
In this case both approaches still have the linear time complexity, however, my approach becomes more complicated with more edge cases and the necessity to update two maps, both not contiguous in the memory.
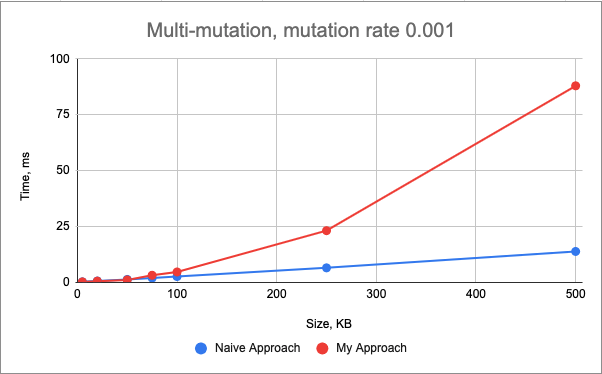
Finally, multi-mutation behaves similar to the insert() mutation, which means that the performance is dominated by the insert() mutation in this case.
🔗 Conclusion
Initially, using change log to only store the differences between the parent genome and the offspring genome seemed like a great idea to improve both the time performance and the memory use of the genome class.
However, careful benchmarking shows that it is very hard to beat the data structures and the algorithms from the standard library. This information (combined with the similar outcomes from my teammates, who developed different algorithms for the change logging) will hopefully be helpful for the future developments of MABE.
My algorithms improves the memory use, because it alleviates the need for deep copy of genome at every generation (i.e., every iteration when the mutations occur) and only stores the mutations instead.
🔗 A brainstorm of potential improvements and optimizations 
There are multiple things in the algorithm that could be optimized, from both algorithms and code perspective. Some of them are:
- As the benchmarking graphs show,
insert()function is currently the bottleneck - optimizing it will result in better performance of the whole genome class - Currently, the reconstruction algorithm will always reconstruct a full offspring genome, which is very inefficient, as sometimes only a part of genome is needed. A couple of improvement could be done here:
- Reconstruct only the sites that are requested
- Use change_log to check if there was a mutation within the requested sites. If not - return a pointer to the requested index in the parent genome
🔗 Acknowledgments
I would like to thank the organizers of the WAVES workshop. It was an extremely valuable experience for me and I learned so much I couldn’t have imagined was possible 10 weeks ago. :exploding_head:
In addition, I would like to thank my mentors Cliff and Jory for giving me the opportunity to work on this super cool and interesting project, as well as for all the brainstorming sessions and for answering my questions and helping throughout the workshop.


Last but not least, I want to thank team MABE for creating friendly and encouraging atmosphere and for always being there when I needed help! I wish everyone success in their studies and career and I hope we will stay in touch! ![]()
![]()
![]()




This work is supported through Active LENS: Learning Evolution and the Nature of Science using Evolution in Action (NSF IUSE #1432563). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.